Project summary:
Heterogeneous catalysis is heavily used in industry and is pivotal in our efforts to make chemical processes greener and more efficient. Direct experimental identification of the catalytically active sites and reaction mechanisms in heterogenous catalysts is challenging, hindering the ability to design catalysts for particular application [1]. Our recent work [2-3] on microporous zeolitic catalysts using biased ab initio molecular dynamics highlighted the fluctional nature of the catalyst surface, challenging the conventional view of these catalysts as being static under solvation. However, achieving such computational results for realistic catalytic systems is either extremely costly [4-5] using ab initio treatments only or is plainly unattainable due to unsuitability/unreliability of available empirical force fields. These are the reasons for the continuing Maddox “scandal”, i.e., that it still remains impossible to predict the structure of even the simplest crystalline solids from first principles based only on a knowledge of their chemical composition.
Therefore, the objectives of the project are:
- Develop machine learning potentials (MLPs), that will accelerate exploration of catalyst configurational space by few orders of magnitude while retaining ab initio accuracy. In addition, the MLPs can be used as a surrogate model enhancing potential energy space sampling using ab initio method, allowing for continuous improvement of MLP via active learning. MLPs are expected to accelerate configurational space exploration by up to 3 orders of magnitude, while adoption of active learning strategies along with multi-Hamiltonian hybrid MC approaches will allow for the discovery of “new chemistry”. We have already made a significant progress along this direction [6].
- Develop data-driven bias-free dimensionality reduction schemes in which the good collective variables accelerating rare events, i.e., chemical reactions, will be selected automatically. Starting with a few known configurations (final catalyst, initial reaction mixture or some stable long-living intermediate), this scheme will allow us to effectively explore optimal low-dimensional representation of catalyst configuration space focusing on the most likely (trans)formation paths. It will enable routine open-ended searches of complex (trans)formation in catalytic systems resolving metastable configurations and overcoming the barriers between them, which are both typically inaccessible to experimental probing
These new tools will enable predicting structures of catalysts from a knowledge of their chemical composition and synthesis conditions. The successful candidate will be able to build on accumulated knowledge base in our group both with respect to development of machine learning potentials and generation of data-driven collective variables.
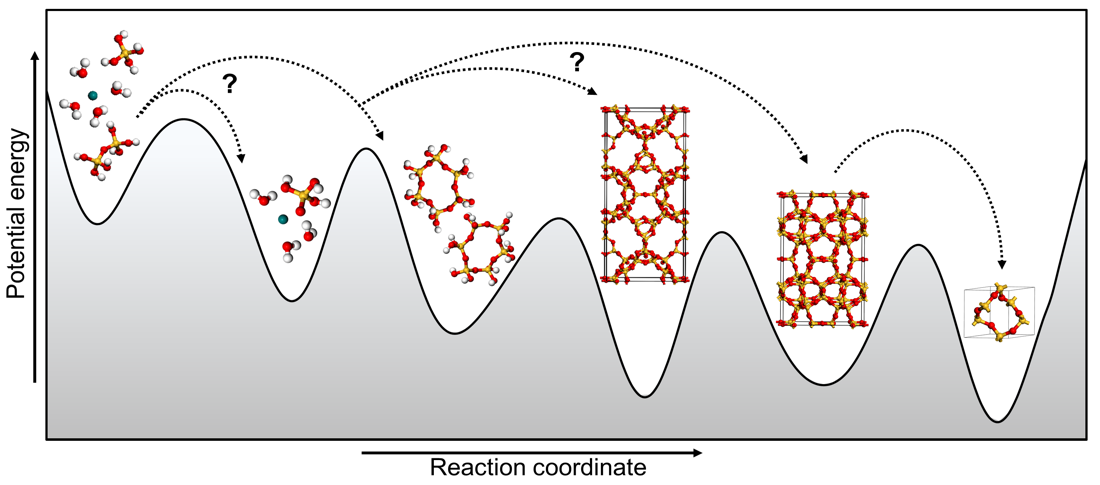
The plan of the project is to showcase this general-purpose approach on an industrially extremely important class of catalysts with a particularly challenging configurational space, the zeolites [7-8]. The grand application challenge will be the realistic atomic-level modelling of zeolite synthesis, a long-standing unresolved problem with many competing hypotheses. By charting the configurational space of transformation process from reaction mixture to a zeolite framework the successful applicant will not only settle the long-standing debate but will open new routes to novel or improved zeolitic materials by modifying existing synthetic procedures.
Profile of a successful candidate:
Required - MSc. or equivalent in Chemistry, Physics, Material Science or a related field; good knowledge of English; experience in programming (ideally Python or similar)
Advantageous, but nor required - background in Machine Learning, Statistics, Statistical Mechanics and Quantum Chemistry/Physics; experience with molecular simulations, high-performance computing (including GPU accelerated one) and Linux.
Relevant publications of the project leader:
[1] Grajciar, L., Heard, C.J., Bondarenko, A.A., Polynski, M.V., Meeprasert, J., Pidko, E.A., Nachtigall, P. (2018): Towards Operando Computational Modeling in Heterogeneous Catalysis. In: Chemical Society Reviews (IF=40.1), 22, 8307-8348
[2] Heard, C. J., Grajciar, L., Nachtigall, P., (2019): The Effect of Water on the Validity of Loewenstein’s Rule. In: Chemical Science (IF=9.063), 10, 5705-5711.
[3] Heard, C.J., Grajciar, L., Rice, C.M., Pugh, S.M., Nachtigall, P., Ashbrook, S., Morris, R.E. (2019): Fast Room Temperature Lability of Aluminosilicate Zeolites. In: Nature Communications (IF=11.9) doi: 10.1038/s41467-019-12752-y.
[4] Grajciar, L. (2015): Low-memory Iterative Density Fitting. In: Journal of Computational Chemistry (IF=3.2), 36, 1521-1535. Cover article of the Issue 20.
[5] Lazarski, R., Burow, A. M., Grajciar, L., Sierka, M., (2016): Density Functional Theory for Molecular and Periodic Systems Using Density Fitting and Continuous Fast Multipole Method: Analytical Gradients. In: Journal of Computational Chemistry (IF=3.2), 37, 2518-2526.
[6] Erlebach, A., Nachtigall, P., Grajciar, L., (2021): Accurate large-scale simulations of siliceous zeolites by neural network potentials. In: arXiv preprint https://arxiv.org/abs/2102.12404.
[7] Roth, W. J., Nachtigall, P., Morris, R. E., Wheatley, P. S., Ashbrook, S. E., Seymour, V., Chlubna, P., Grajciar, L., Polozij, M., Zukal, A., Shvets, O., Cejka, J. (2013): A Family of Complex Zeolites with Controlled Pore Size Prepared Through a ‘top down’ Method. In: Nature Chemistry (IF=26.2), 5, 628-633.
[8] Heard, C. J., Grajciar, L., Uhlik, F., Shamzy, M., Opanasenko, M., Cejka, J. Nachtigall, P., (2020): Zeolite (In)Stability under Aqueous or Steaming Conditions. In: Advanced Materials (IF=27.4), 32, 2003264.
Current research grants of the project leader:
Junior group leader research project Primus/20/SCI/004 of Charles University (two postdocs and one PhD student) - “Computational catalysis goes operando”, principal investigator, total budget € 250.000, (2020-2022)
Deadline is closed
